What causes Cystic Fibrosis?

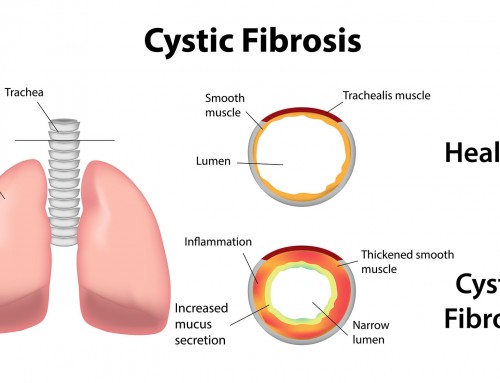
Cystic fibrosis is caused by mutations in the CFTR gene. The CFTR gene is responsible for encoding the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) protein. This protein primarily functions as a chloride channel that regulates the flow of ions across the cell membrane, affecting water osmosis and the subsequent composition of mucus and sweat. The CFTR protein can also regulate other channels across cell membranes (i.e. sodium ions), which are essential for the proper functioning of organs such as lungs and pancreas. The absence or alteration of CFTR protein disrupts the function of the ion channel. This causes production of thick, sticky mucus which traps bacteria, and cannot be cleared away from airways and ducts, leading to the signs and symptoms of cystic fibrosis.
To date, more than 1000 mutations in the CFTR gene have identified. The severity of the symptoms depends on the type of mutation in the CFTR gene. There are five general classes of CFTR mutations, which differ by the mechanism in how the CFTR function is disrupted. In general, Class I to Class III mutations are loss-of-function mutations, and these classes present more severe symptoms of cystic fibrosis. Class IV to Class V mutations result in reduced function, and produce milder effects.
Class I mutations
This class affects the biosynthesis of the protein, resulting in a shortened protein. E.g. G542X, R553X.
Class II mutations
Delta F508, a mutation belonging to this class, is the most common mutation that caused cystic fibrosis. This class of mutations affects protein maturation and folding, resulting in non-functional protein that fail to reach the cell membrane.
Class III mutations
This class affects chloride ion channel regulation, resulting in proteins that are transported to the cell membrane, but do not respond to stimulation. An example of this class is the missense mutation G551D.
Class IV mutations
This class affects chloride ion conductance, resulting in a protein that is transported to the cell membrane and responds to stimuli, but generates a reduced ion current. The protein channel does not open properly, but is still able to maintain partial function. E.g. R117H.
Class V mutations
This class affects protein stability, resulting in a protein channel that is inherently unstable due to incorrect splicing of the CFTR gene. E.g. Q1412X.
References:
Castellani C, Cuppens H, Macek M et al. (2008). Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. Journal of Cystic Fibrosis. 7: 179-196.
Richards CS, Bradley LA, Amos J et al. (2002). Standards and Guidelines for CFTR Mutation Testing. Genetics In Medicine. 4(5): 379-391.
Watson MS, Cutting GR, Desnick RJ et al. (2004). Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genetics In Medicine. 6(5): 387-391.
DNA In the News2017-04-06T20:44:10+00:00