What is Cystic Fibrosis?

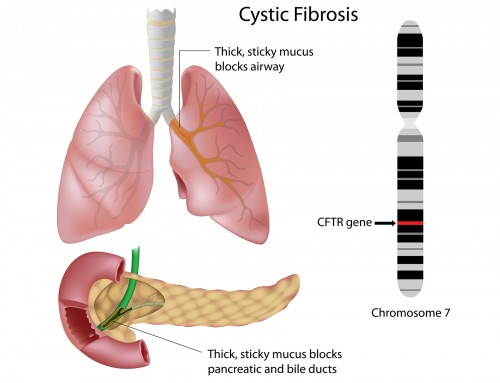
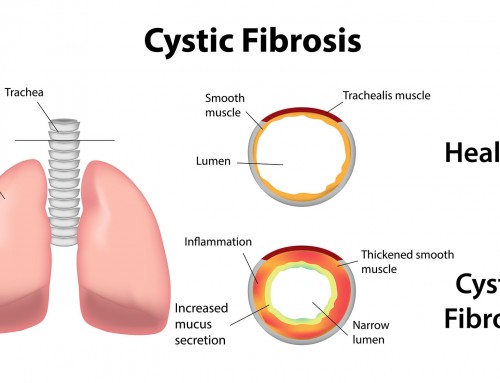
Cystic fibrosis is a genetic disorder characterized by production of abnormally thick sticky mucus, affecting primarily the respiratory and digestive system.
Mucus is a slippery secretion that hydrates and protects the linings of body cavities and other internal organs. The thick and sticky mucus produced in people with cystic fibrosis has poor mucociliary clearance, and accumulates in the body over time. This abnormal mucus can obstruct airways and ducts, leading to chronic obstructive lung disease, recurrent bacterial infections, breathing difficulties and digestive problems. The signs and symptoms of the disease vary in severity. Early diagnosis and treatment for cystic fibrosis is recommended, as the disease can lead to life-threatening complications if left untreated.
Cystic fibrosis is an autosomal recessive disorder caused by mutations in the CFTR (Cystic Fibrosis Transmembrane conductance Regulator) gene. Mutations need to be present in both copies of the gene for an individual to inherit the disease. If only one copy of mutated CFTR gene is inherited, individuals are considered carriers of cystic fibrosis and appear normal. The CFTR gene encodes the protein which forms a channel in regulating the flow of chloride ions and water across cell membranes. Absence or abnormality of the CFTR protein disrupts the function of the chloride channel, resulting in the production of abnormally thick mucus which leads to the characteristics of cystic fibrosis.
To date, there is no cure for cystic fibrosis but there are different treatments available to ease or prevent symptoms. For example, antibiotics are used to treat recurrent lung infections. Airway clearance techniques can be used for preventing mucus buildup in the respiratory system. Vitamin supplementation and pancreatic enzyme tablets can be taken to help with digestion problems. Diet and exercise is also important to improve lung function and ventilation, thereby increasing the life expectancy of affected people with cystic fibrosis.
References:
Castellani C, Cuppens H, Macek M et al. (2008). Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. Journal of Cystic Fibrosis. 7: 179-196.
Richards CS, Bradley LA, Amos J et al. (2002). Standards and Guidelines for CFTR Mutation Testing. Genetics In Medicine. 4(5): 379-391.
Watson MS, Cutting GR, Desnick RJ et al. (2004). Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genetics In Medicine. 6(5): 387-391.
DNA In the News2017-04-06T20:44:10+00:00